2022年(nián)2月10日,FDA召開(kāi)ODAC會議(yì)審評信達生物(wù)與禮來制藥的PD-1抗體信迪利單抗。

專家委員(yuán)會的最終投票結果為(wèi)14:1,認為(wèi)信迪利單抗應該補充臨床試驗才能(néng)獲得批準。一(yī)般情況下(xià),FDA會遵照ODAC專家委員(yuán)會的投票結果。

禮來對這(zhè)一(yī)結果表示失望,原本希望通(tōng)過信迪利單抗可(kě)以通(tōng)過激進的價格策略影響美國(guó)的醫(yī)療系統,而讓患者獲益。

FDA的問(wèn)題集中在三點,第一(yī)是臨床格局的變化,第二是單國(guó)臨床研究數(shù)據是否适用于美國(guó)患者,第三是PFS終點是否足夠。
對于第一(yī)點,FDA認為(wèi)2018年(nián)8月20日Keytruda聯合化療即獲批用于治療非鱗狀NSCLC,ORIENT-11在此之後才啓動,臨床一(yī)線療法已經從(cóng)化療變為(wèi)免疫治療+化療聯合治療。
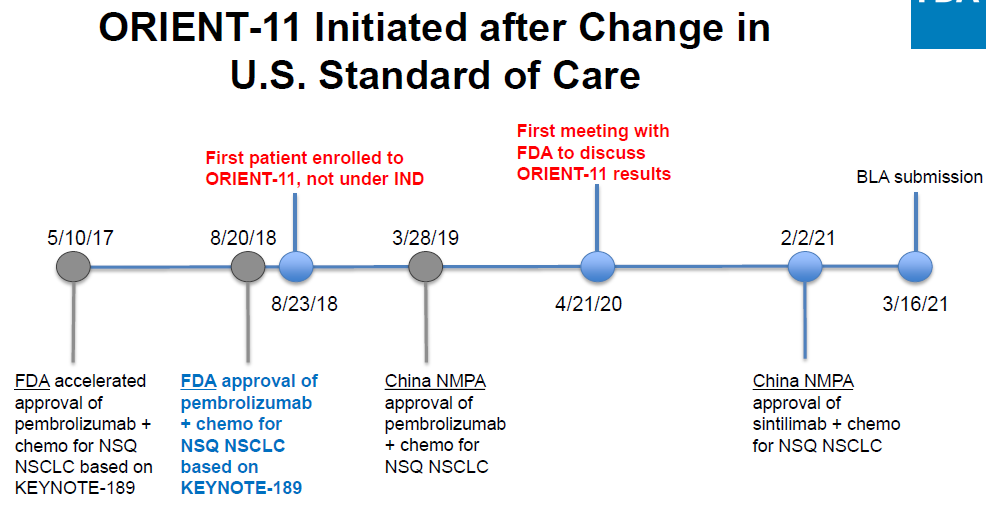
對于第二點,FDA認為(wèi)中國(guó)數(shù)據不足以申報用于治療美國(guó)患者。


對于第三點,美國(guó)批準NSCLC基本都(dōu)以OS終點為(wèi)主,信迪利單抗以PFS申報是否足夠。
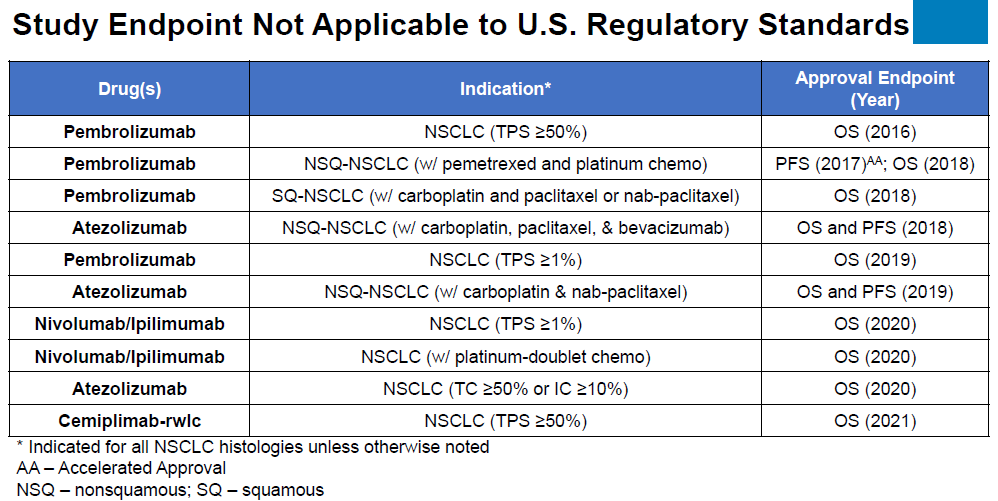
FDA強調,審評中不會考慮藥品價格因素。

信達生物(wù)與禮來制藥則針對性的給出回應,認為(wèi)中美梁果對于非鱗狀NSCLC的診斷、分期和(hé)治療格局都(dōu)是類似的,ORIENT-11中期分析達到PFS終點,OS分析也表明(míng)達到了(le)有意義的臨床獲益,臨床标準是類似的且PK/PD對于種族因素不敏感,因此臨床數(shù)據适用于美國(guó)患者。

FDA與信達/禮來的結論對立鮮明(míng)。

FDA認為(wèi)在某些具體問(wèn)題上(shàng),申請(qǐng)者缺少(shǎo)必要(yào)的溝通(tōng),如(rú)臨床方面方面,如(rú)果實現與FDA進行(xíng)溝通(tōng),後者可(kě)能(néng)會建議(yì)采取與已上(shàng)市PD-1+化療的頭對頭對照,并采用OS終點。

此外(wài),FDA直截了(le)當地(dì)引用中國(guó)藥監部門的報告,80%的中國(guó)臨床研究存在欺詐或者不夠标準。

投票問(wèn)題為(wèi)是否需要(yào)補充額外(wài)的臨床研究,以證明(míng)适用于美國(guó)患者和(hé)美國(guó)的臨床實踐。最終投票結果15位專家中14位都(dōu)認為(wèi)應該補充臨床試驗。

總結
如(rú)前文所述,此次審評的挑戰更多是源于FDA在新形勢下(xià)的政策調整,也是未來更多國(guó)産創新藥出海(hǎi)要(yào)面臨的挑戰。ODAC最終以14:1的投票結果建議(yì)拒絕批準信迪利單抗,要(yào)求補充臨床試驗,意味着中國(guó)創新藥出海(hǎi)需要(yào)适應新的規則。信達生物(wù)作(zuò)為(wèi)先行(xíng)者,其探索與努力值得尊敬,也終将吹響中國(guó)創新藥全面進軍國(guó)際市場的号角。